

Virtual Drug Screening: Bekannte Medikamente stoppen das Virus in der Simulation
Von 42.000 Wirkstoffen hemmen zehn den Covid-19-Erreger in Computersimulationen effektiv. Darunter sind vier zugelassene Hepatitis-C-Präparate.

(Bild: Stefan F. Sämmer/Uni Mainz)
Auf der Suche nach einem Wirkstoff gegen das neue Corona-Virus SARS-CoV-2 erkannte ein Programm auf dem Supercomputer Mogon II an der Mainzer Universität eine Reihe interessanter Wirkstoffe. Per Virtual Drug Screening hatte sich ein Team um Professor Dr. Thomas Efferth in Simulationen auf die Suche nach einem Heilmittel begeben. Außerhalb von Pandemien nutzt Efferth die Screening-Verfahren, um Wirkstoffkandidaten gegen Krebs zu identifizieren. Mitte Januar startete er sein Anti-Corona-Projekt.
Rund 42.000 Substanzen bezogen die Forscher in ihre Suche ein, darunter Naturstoffe, experimentelle Wirkstoffe und 1577 zugelassene Präparate aus einer FDA-Bibliothek (Food and Drug Administration, USA). Für alle berücksichtigten Wirkstoffmoleküle lagen deren räumliche Strukturen als 3D-Daten im SDF-Format vor (Structure Data File).
Molukulares Docking
Als Gegenstück wählten die Forscher drei vielversprechende Proteine im Corona-Virus aus. Für jeden einzelnen Wirkstoff erprobten sie virtuell die molekularen Docking-Möglichkeiten und wie stark Wirkstoffmolekül und Virusprotein aneinander binden. Die Stärke einer Bindung kann man sich dabei wie bei einem Puzzleteil vorstellen: Je genauer die Formen ineinandergreifen, desto harmonischer fügt sich das neue Teil ein und desto schwerer lässt es sich wieder herausfummeln. Beim molekularen Docking spielen außer den räumlichen Strukturen auch elektrostatische Wechselwirkungen eine bedeutende Rolle.

Die Bindekraft ist entscheidend, wie Molekularbiologe Efferth erläutert. Das hängt mit der Funktion der ausgewählten Virusproteine zusammen. Eines davon, das sogenannte Spike-Protein, bildet zum Beispiel die spitzen Strukturen auf der Virusoberfläche, die dem Virus seinen Namen „Corona“ geben und mit denen sich der Keim an Rezeptoren von menschlichen Lungenzellen ankoppelt. Sobald sich ein Wirkstoff an diesem Protein anlagert und dadurch den Befall der Lunge abblockt, kann das die Infektion beim Menschen verhindern.
(Bild: Onat Kadioglu/Thomas Efferth )
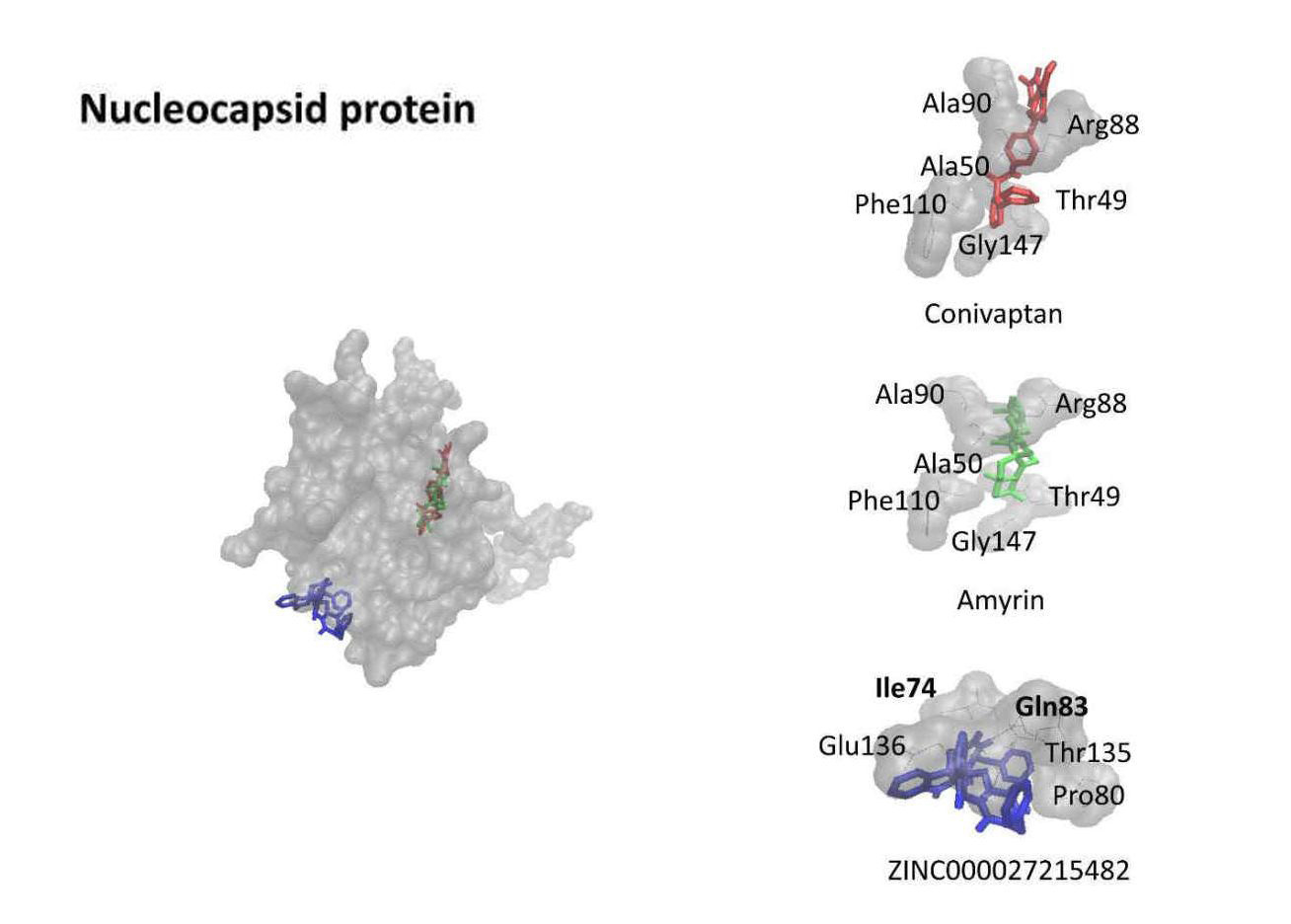
Ein weiterer Virenbaustein, das Nucleocapsid-Protein, dient dem Aufbau einer Schutzkapsel für das Virus-Erbgut. Eine Wirksubstanz, die sich mit diesem Protein verbindet, könnte effektiv die Zusammensetzung neuer Viren im menschlichen Körper blockieren. Das dritte Protein ist wichtig für den Stoffwechsel des Erbguts. Ein Wirkstoff, der diesen Prozess hemmt, stoppt die Virenvermehrung.

(Bild: Onat Kadioglu/Thomas Efferth )
Puzzlespiel mit Protein
Die Forscher gingen etappenweise vor. Sie nutzten die Open-Source-Software-Umgebung PyRx, die alle Schritte von der Datenaufbereitung bis zur Ergebnisanalyse unterstützt. Für das Vor-Screening der Wirkstoffe verwendeten sie das in PyRx eingebundene Programm AutoDock VINA, ebenfalls Open Source. Bereits dieses Tool probiert für jeden Wirkstoff in silico das molekulare Docking auf der gesamten Proteinoberfläche aus. Es vergleicht die räumlichen Strukturen und errechnet die Bindungskraft. Mit diesem ersten Tool ermittelten die Mainzer für jedes der drei betrachteten Virusproteine 100 erfolgversprechende Wirkstoffe.
Die Bindungskraft von diesen 300 Favoriten ermittelte ein weiterer Simulationsdurchlauf mit AutoDock 4.2 noch einmal gründlicher. AutoDock verwendet wie AutoDock VINA für die Darstellung der Molekularstruktur das Dateiformat PDBQT. Das im Vergleich zu VINA exaktere Tool erfordert etwa um zwei Größenordnungen mehr Rechenzeit für das molekulare Docking. Es untersucht dabei aber sämtliche denkbaren Andockstellen für die Wirkstoffmoleküle und bezieht auch unterschiedliche Stellungen und Torsionen von Seitenketten im Molekülaufbau mit ein.
AutoDock 4.2 verwendet Approximationsalgorithmen und bestimmt damit die Bindungskraft zwischen Wirkstoffmolekül und Eiweiß näherungsweise. Daher ließen die Mainzer ihre Berechnungen im Supercomputer millionenfach durchlaufen, um Mittelwerte und Standardabweichungen zu ermitteln. Letztlich erforderten die virtuellen Docking-Versuche über 30 Milliarden Einzelberechnungen.
Eine zusätzliche Untersuchung der vielversprechenden Anti-Corona-Wirkstoffe betraf Eigenschaften, die für Medikamente essenziell sind, zum Beispiel die Wasserlöslichkeit, die Molekülgröße oder die Existenz bestimmter funktionaler Bausteine. Hier half künstliche Intelligenz und der Aufbau neuronaler Netze, die Wirksamkeit der vielversprechenden Substanzen vorherzusagen. Die Wissenschaftler trainierten neuronale Netze mit funktionierenden Präparaten, die ähnlich arbeiten wie die angestrebte Wirkweise. Wirkstoffe gegen das Spike-Protein beispielsweise verglich eine KI mit Medikamenten, die ebenfalls Viren am Andocken an menschlichen Zellen hindern.
Sofort einsetzbar
Die Ergebnisse lassen aufhorchen. Die Mainzer haben in ihren Computersimulationen nicht nur sieben vielversprechende Wirkstoffe gegen das Corona-Virus entdeckt. Einer dieser Wirkstoffe griff sogar zwei der betrachteten Virusproteine an. Insbesondere befinden sich unter den Favoriten gegen das Spike-Protein vier zugelassene Hepatitis-C-Medikamente namens Simeprevir, Paritaprevir, Grazoprevir und Velpatasvir.
Durch ihre Wirkweise könnten sie in der Lage sein, Ansteckungen mit Covid 19 sowie dessen Ausbreitung in der Lunge zu verhindern. „Bereits zugelassene Medikamente dürfen Ärzte im Rahmen eines individuellen Heilversuchs auf eigene Verantwortung einsetzen“, erklärt Efferth. Das sei eine große Chance, jetzt schnell gegen die Krankheit vorzugehen. Efferth hat nach eigenen Angaben Kontakt zu Ärzten, die nun diesen Weg in Betracht ziehen.
Simuliert statt geraten
Der Ansatz, bereits zugelassene Medikamente gegen Covid-19 einzusetzen, ist nicht neu. So war bereits das Malariamittel Chloroquin im Gespräch, von dem bekannt ist, dass es antivirale Wirkung aufweist. Außerdem brachten Mediziner schon früh das Ebola-Medikament Remdesivir ins Gespräch. Bei Ebola besteht das Genom wie bei SARS-CoV-2 aus Ribonukleinsäure (RNA-Virus). Zudem gab es bereits in der ersten SARS-Epidemie 2002/2003 Hinweise auf eine gewisse Wirkung gegen Corona-Viren. Diese ersten Vorschläge entstanden allerdings noch ohne fundierte Ergebnisse breiter molekularer Docking-Versuche in Computersimulationen.
Klinische Studien unerlässlich
Als ein weiterer heißer Kandidat gegen Covid 19 hat sich in der Simulation ein Naturstoff aus dem Japanischen Geißblatt (Lonicera japonica) erwiesen. Die im Supercomputer identifizierten Wirkstoffe müssen allerdings zunächst in Laborexperimenten und klinischen Studien bestehen. Aber auch ihre Entdeckung ist ein großer Zeitgewinn. Üblicherweise umfasst der Entwicklungsprozess eines neuen Medikamentes fünf bis zehn Jahre. „In unserem Fall haben wir eine Vorauswahl getroffen und sparen uns die Laborversuche bis zu dem Punkt, wo es zu Tierversuchen kommt“, erläutert Efferth. Damit sind schon Jahre an Forschung eingespart.
Zur Vermeidung von Tierversuchen ist das molekulare Docking in der Computersimulation nicht geeignet. Die Simulation der Bindung zwischen Wirkstoff und Erregerprotein sagt noch nichts über dessen Wirkung im Körper aus. Was in der Verdauung passiert und insbesondere im Leberstoffwechsel, das lässt sich mit dem hier geschilderten Verfahren nicht simulieren.
Im Fall der Hepatitis-Medikamente kann die Zulassung als Covid-19-Mittel noch deutlich schneller erfolgen, weil die Sicherheit der Medikamente bereits erwiesen ist. Allerdings wäre noch eine klinische Studie notwendig, um zu belegen, ob die Mittel tatsächlich gegen das Virus SARS-CoV-2 wirken. Schon nach sechs bis zwölf Monaten könnten die Behörden die Mittel dann als Covid-19-Medikament freigeben.
Ihre Untersuchungsergebnisse haben die Forscher auf der Website der Weltgesundheitsorganisation (WHO) veröffentlicht.
Dieser Artikel stammt aus c't 13/2020.
(agr)
